








Chemická analýza struktury povlaků s rozlišením 170 nm
Rubrika: Povrchová ochrana
V současnosti je zažitá představa, že rozlišení prvkové EDS analýzy na elektronových mikroskopech činí cca 2 μm. Při užití běžných postupů na mikroskopech s wolframovým vláknem je to pravda, neboť při těchto měřeních je obvykle užíváno urychlovacího napětí 20 kV.
Uvažujeme-li hustotu zinku 7,14 g/cm3 a hustotu železa 7,87 g/cm3 [1]. Intermetalické fáze soustavy Fe-Zn se pohybují v rozmezí koncentrací 70 – 94 % pro jednoduchost tedy uvažujme střední hustotu intermetalik jako vážený průměr hustoto obou kovů, s vahou Zn = 0,85 a Fe = 0,15. Získáme hodnotu 7,25 g/cm3 (zdůrazněme, že přesná hodnota hustot intermetalik nyní není důležitá).
V případě materiálů s vysokou hustotou (tedy kovů) se tvar interakčního objemu mění z hruškovitého na polokulovitý[ 2], lze tedy říci že průměr interakčního objemu je přibližně dvojnásobkem jeho hloubky.
Při užití zjednodušeného vztahu pro hloubku interakčního objemu h[μm] =(E1,5 × 0,1)/ρ , kde ρ je hustota [g/cm3] a E je urychlovací napětí [kV] získáme hloubku interakčního objemu 1,23 μm. Půdorys interakčního objemu má potom skutečně průměr 2,46 μm.
Nabízí se otázka, zda lze snížit hodnotu urychlovacího napětí. V případě běžných elektronových mikroskopů tím značně klesne proud svazku a tedy i intenzita signálu, zhorší se kvalita snímku a na některých pracovištích tento zásah může vyvolat i problémy kvantifikace díky nedostupnosti kalibrace.
Nejdůležitějším momentem však je poloha běžně užívaných Kα spektrálních čar. Pro zinek má hodnotu 8,637 keV a pro železo 6,403 keV [1]. Pro buzení těchto čar je nutné mít urychlovací napětí nastaveno minimálně na tutéž hodnotu, aby čára byla vůbec pozorovatelná. Běžná praxe [2] doporučuje dvoj- až trojnásobek hodnoty za účelem správné kvantifikace. Je tedy zřejmé, že při kalibraci podle K‑čar je užití menšího napětí než 15 kV, nebo dokonce 10 kV, nemyslitelné.
Při užití čar Lα (Fe: 0,705 keV, Zn: 1,012 keV[1]) vyvstává problém s kalibrací a zejména s překryvy s čarami ostatních prvků (O, C, Na, Sb, a další) v této nízkoenergetické oblasti, což vyžaduje vysoké spektrální rozlišení použité aparatury.
Když už přichází v úvahu možnost měření s užitím čar série L, je třeba překonat problémy s povrchovou kontaminací organickými látkami (i při sebelepším čištění vzorku k ní dochází kvůli kontaminaci z vakuového systému mikroskopu) a také nevyhnutelnou oxidací vzorku.
Naše pracovitě je vybaveno elektronovým mikroskopem se polem řízeným zdrojem elektronů (fieldemissiongun, FEG) a velmi solidní EDS aparaturou (pološířka čáry (FWHM) Kα Mn činí maximálně 127 eV). FEG zdroj umožňuje dosahovat vysokých proudů svazku a kvalitního obrazu i při velmi nízkých urychlovacích napětích.
Systém EDS analýzy umožňuje kalibraci při nejméně 4 kV. Kontaminace uhlíkem je odstraňována plazmatickým čistícím systémem a při řádné přípravě vzorků a standardu není problém nevyhnutelnou oxidaci zahrnou do kalibrace. Kalibračním standardem jsou monokrystaly intermetalické sloučeniny FeZn13 (tzv. ζ – fáze), která má velmi úzký interval homogenity při složení 92,86 % at. Zn + 7,14 % at. Fe (v hmotnostních procentech je to 93,84 % Zn a 6,16 % Fe).
Při užití urychlovacího napětí 4 kV jsou splněny excitační požadavky pro obě L – čáry, při řádné kalibraci a statistickém zpracování lze dosáhnout přesnosti analýz ±0,1 %. Pomocí uvedeného zjednodušeného vztahu vychází hloubka interakčního objemu 0,11 μm. Vzhledem k tomu, že urychlovací napětí i energie obou čar
jsou již velmi nízké, je vhodné pro výpočet použít přesnějšího Anderson – Haslerova vztahu [3], který uvažuje rozdíl mezi hranou spektrální série a energií budícího svazku elektronů. Tímto vztahem získáme hloubku, ze které vychází signál zinku 81,5 nm a pro železo je to 85,7 nm. Za předpokladu polokulového interakčního objemu (viz výše) činí tedy rozlišení při měření koncentračního profilu maximálně 170 nm.
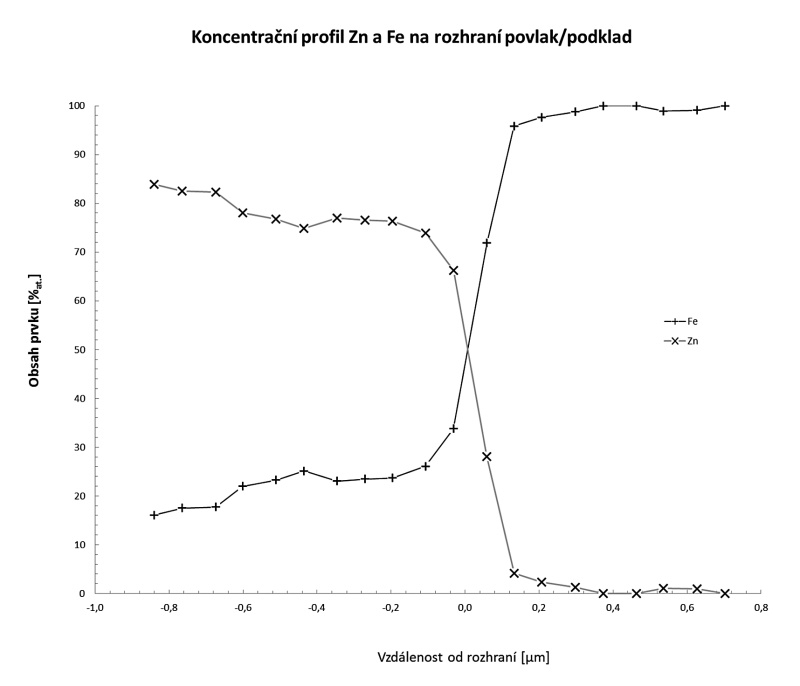
Uvedeno metodou je tedy možné spolehlivě zkoumat koncentrační profil vrstev tlustých menších, než 1 μm, jako je tomu např. u fáze Γ a Γ1 na rozhraní žárově zinkovaného povlaku a podkladové oceli (obr. 1).
LITERATURA:
[1] Richtera, L.: Periodický systém prvků. VŠCHt, Praha, 2013.
[2] Goldstein J. et al.: ScanningElectronMicroscopy and X-rayMicroanalysis.s. 286. Springer, New York 2007.
[3] Anderson, C.A., Hasler, M.F.: Proceedingsofthe 4th International Conference on X-ray Optics and Microanalysis (R. Castaign, P. Deschamps and J. Phillibert, eds.), s. 310. Hermann, Paris 1966.
NEJčtenější souvisejicí články (v posledních 30-ti dnech)
Na základě poptávky našich zákazníků na maskování částí ocelových konstrukcí před žárovým pozinkováním jsme se začali za...
Ocel je moderní stavební materiál, který má široké možnosti uplatnění ve všech typech staveb. Z hlediska požární odolnos...
NEJlépe hodnocené související články
Objednatele žárového pozinkování mnohdy znepokojuje různorodý vzhled povlaku. U zakázek provedených z rozmanitého materi...
K tomuto článku bola zvolená téma osvetľujúca skúsenosti a prax investorov z radov energetiky, využívajúcich služieb sie...
Korozivzdorné oceli patří mezi konstrukční materiály s vysokou korozní odolností v závislosti na způsobu jejich legování...
NEJdiskutovanější související články
Na základě poptávky našich zákazníků na maskování částí ocelových konstrukcí před žárovým pozinkováním jsme se začali za...
Přelom června a července letošního roku bude ve znamení Mistrovství světa ve fotbale 2010. Tuto sportovní událost poprvé...



ISSN 1803-8433 | © Copyright 2002 - 2026 KONSTRUKCE Media, s.r.o.
Jakékoliv užití obsahu včetně převzetí, šíření či dalšího zpřístupňování článků a fotografií je bez souhlasu nakladatelství zakázáno.
KONSTRUKCE Media, s.r.o., se sídlem Starobělská 1133/5, 700 30 Ostrava, zapsaná v obchodním rejstříku vedeným u Krajského soudu v Ostravě, oddíl C, vložka 22003, Identifikační sídlo: 25851276 | Tel.: 597 317 578 | Fax: 597 579 166


